
Abstract
BACKGROUND
The glucocorticoid dexamethasone prevents nausea and vomiting after surgery, but there is concern that it may increase the risk of surgical-site infection.
METHODS
In this pragmatic, international, noninferiority trial, we randomly assigned 8880 adult patients who were undergoing nonurgent, noncardiac surgery of at least 2 hours’ duration, with a skin incision length longer than 5 cm and a postoperative overnight hospital stay, to receive 8 mg of intravenous dexamethasone or matching placebo while under anesthesia. Randomization was stratified according to diabetes status and trial center. The primary outcome was surgical-site infection within 30 days after surgery. The prespecified noninferiority margin was 2.0 percentage points.
RESULTS
A total of 8725 participants were included in the modified intention-to-treat population (4372 in the dexamethasone group and 4353 in the placebo group), of whom 13.2% (576 in the dexamethasone group and 572 in the placebo group) had diabetes mellitus. Of the 8678 patients included in the primary analysis, surgical-site infection occurred in 8.1% (354 of 4350 patients) assigned to dexamethasone and in 9.1% (394 of 4328) assigned to placebo (risk difference adjusted for diabetes status, −0.9 percentage points; 95.6% confidence interval [CI], −2.1 to 0.3; P<0.001 for noninferiority). The results for superficial, deep, and organ-space surgical-site infections and in patients with diabetes were similar to those of the primary analysis. Postoperative nausea and vomiting in the first 24 hours after surgery occurred in 42.2% of patients in the dexamethasone group and in 53.9% in the placebo group (risk ratio, 0.78; 95% CI, 0.75 to 0.82). Hyperglycemic events in patients without diabetes occurred in 22 of 3787 (0.6%) in the dexamethasone group and in 6 of 3776 (0.2%) in the placebo group.
CONCLUSIONS
Dexamethasone was noninferior to placebo with respect to the incidence of surgical-site infection within 30 days after nonurgent, noncardiac surgery. (Funded by the Australian National Health and Medical Research Council and others; PADDI Australian New Zealand Clinical Trials Registry number, ACTRN12614001226695
Methods
TRIAL DESIGN
In this pragmatic, international, multicenter, randomized, placebo-controlled, triple-blind (patient, anesthesiologist, and assessor), noninferiority trial, patients were assigned to receive 8 mg of intravenous dexamethasone or matching placebo after induction of general anesthesia and before surgical incision. The rationale and design of the trial have been reported previously.11 The trial and the protocol (available with the full text of this article at NEJM.org) were approved by the institutional review board at each participating site.
PATIENT SELECTION AND RANDOMIZATION
Eligible adult patients had an American Society of Anesthesiologists (ASA) physical status classification of I to IV (on a scale of I to VI, with higher classes indicating more severe systemic disease) and were undergoing elective or expedited nonurgent, noncardiac surgery with general anesthesia and an expected operative duration of at least 2 hours as well as an expected postoperative hospital stay of at least 1 night. Patients were excluded if they were undergoing surgery that was time critical, that involved an expected total incision length of 5 cm or shorter, that was associated with a primary infection (e.g., infection related to a prosthesis), or that required the use of intraoperative dexamethasone. Patients were also excluded if they had poorly controlled diabetes mellitus (defined as a glycated hemoglobin level of >9.0%). Eligible patients were randomly assigned in a 1:1 ratio by means of a Web-based service to receive dexamethasone or placebo in random permuted blocks of size 6 and 12, stratified according to trial center and a diagnosis of diabetes mellitus.
TRIAL PROCEDURES
Dexamethasone or placebo (supplied in a 2-ml vial and labeled with a randomization letter) was administered as an intravenous bolus within 5 minutes after induction of anesthesia by the attending anesthesiologist. All other aspects of patient care (including prophylactic antibiotic agents, management of blood glucose levels, and medication for patients with diabetes) followed local protocols and established guidelines. Nontrial glucocorticoids were prohibited for up to 30 days after the index surgical procedure.
The primary outcome was assessed in the modified intention-to-treat, per-protocol, and as-treated populations. The primary analysis was performed in the modified intention-to-treat population, which excluded patients who did not undergo eligible surgery (i.e., surgery with a total incision length of >5 cm when the patient was under general anesthesia), who withdrew consent or whose clinician withdrew the patient from the trial, who could not receive dexamethasone or placebo because they were not available at the center, or who met other exclusion criteria. The per-protocol population included the same criteria as those for the modified intention-to-treat population and further excluded patients who did not receive dexamethasone or placebo or who received open-label dexamethasone (or other glucocorticoid) either intraoperatively or within 30 days postoperatively. All secondary and tertiary outcomes were assessed in the modified intention-to-treat population only.
BLINDING, DATA QUALITY, AND SAFETY
The members of the clinical end-point committee, who were unaware of trial-group assignments, adjudicated all primary-outcome events. A list of committee members and details regarding the monitoring of data quality and site audits are provided in the Supplementary Appendix, available at NEJM.org. Two interim analyses were conducted (after 3379 and 5380 patients had been enrolled) by the independent data and safety monitoring committee, and the trial proceeded to completion.
ETHICS, GOVERNANCE, AND FUNDING
Written informed consent was obtained from all participants. Alfred Health oversaw the trial. The members of the steering committee designed the trial, gathered and analyzed the data, and vouch for the accuracy and completeness of the data and adherence of the trial to the protocol. The writing committee wrote the first draft of the manuscript, and the authors, some of whom were members of the steering committee, prepared the manuscript and made the decision to submit it for publication. There was no commercial involvement in the trial. The statistical analysis plan is available with the protocol at NEJM.org.
OUTCOMES
The primary outcome was the occurrence of a surgical-site infection within 30 days after surgery, determined according to the Centers for Disease Control and Prevention definitions, which comprise three categories: superficial incisional, deep incisional, and organ-space infection (Tables S1 and S2 in the Supplementary Appendix).12 Secondary outcomes included superficial, deep, and organ-space infections within 30 days after surgery, assessed separately; deep and organ-space infections within 90 days after surgery in patients who had insertion of prosthetic material, considered separately; other infections (including urinary tract infections, pneumonia, catheter-related infections, and sepsis) within 30 days after the index procedure; the quality of recovery (as assessed with the use of the 15-item quality-of-recovery [QoR-15] scale, with scores ranging from 0 to 150 and higher scores indicating better quality of recovery) on days 1 and 3013; chronic postsurgical pain at 6 months after surgery; and death or persistent new-onset disability within 6 months after surgery.14 Details regarding tertiary outcomes, adverse events, and safety outcomes (including myocardial infarction, cerebrovascular accident, deep venous thrombosis or pulmonary embolism, and other serious adverse events) are provided in the Supplementary Appendix.
Patients were followed in the postanesthesia care unit, on the first 3 postoperative days, at hospital discharge, and at 30 days and 6 months after surgery. Active postoperative surveillance of surgical-site infection involved several processes. A wound assessment was performed on day 3 if the patient was still hospitalized. On day 30 and at 6 months, patients completed a telephone interview for wound assessment, and their medical records were reviewed to identify the occurrence of trial outcomes. Research staff members collated source documentation to enable outcome adjudication. Scores on the QoR-15 scale were assessed on days 1 and 30.13 Details regarding the outcome adjudication process and assessments (including additional assessments of pain and disability that are not included in this article) are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
A noninferiority margin of 2.0 percentage points was determined with the assistance of clinical experts and was set on the basis of a modified Delphi process and an anticipated incidence of surgical-site infection of 9%.15 With an expected incidence of infection of 9% in each group, we determined that 4303 patients per group would be needed to give the trial 90% power to detect noninferiority of dexamethasone; noninferiority would be concluded if the upper boundary of the two-sided 95% confidence interval for the difference in infection rates was less than 2.0 percentage points. As prespecified in the trial protocol, two interim analyses were planned — when one third and two thirds of the total number of patients had been enrolled. The analyses would use two-sided repeated asymmetric confidence intervals to preserve an overall confidence level of 95% and would use the O’Brien–Fleming function to determine the upper boundary of the confidence intervals and the power-spending function for the lower boundaries. The 90% power using these confidence intervals was confirmed by means of numerical simulation. Target recruitment was set at 8880 patients to account for a 3% loss to follow-up. An overall 5% significance level was used, and no correction for multiple testing was applied, apart from adjustment for the multiplicity of interim analyses for the primary outcome.
The absolute difference in 30-day infection rates was estimated with the use of binomial regression with an identity-link function, with adjustment for diabetes status; a risk ratio was also calculated using a logarithmic-link function. The analyses used 95.6% asymmetric confidence intervals based on the actual timing of the two interim analyses and spending functions. Noninferiority P values were calculated as one-sided, based on a null hypothesis of inferiority, with a significance level of 2.37% to account for the interim analyses.16 Sensitivity analyses were performed to account for missing data by imputing outcomes under “worst–best” and “best–worst” case scenarios (Table S10).17
Comparisons between groups for secondary and tertiary outcomes were estimated as risk ratios (for binary outcomes), differences in medians (for continuous outcomes), and hazard ratios (for time-to-event outcomes). Information regarding all statistical analyses is provided in the statistical analysis plan.
Results
PATIENT ENROLLMENT AND FOLLOW-UP
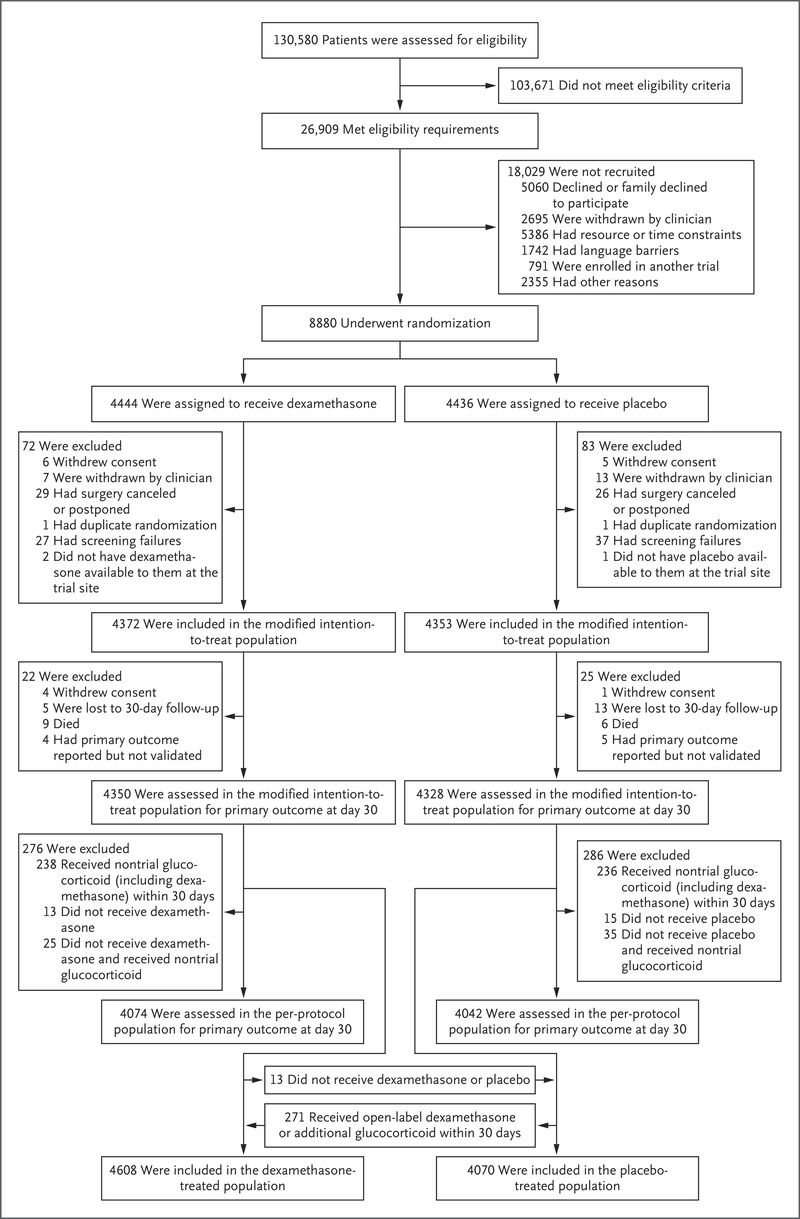
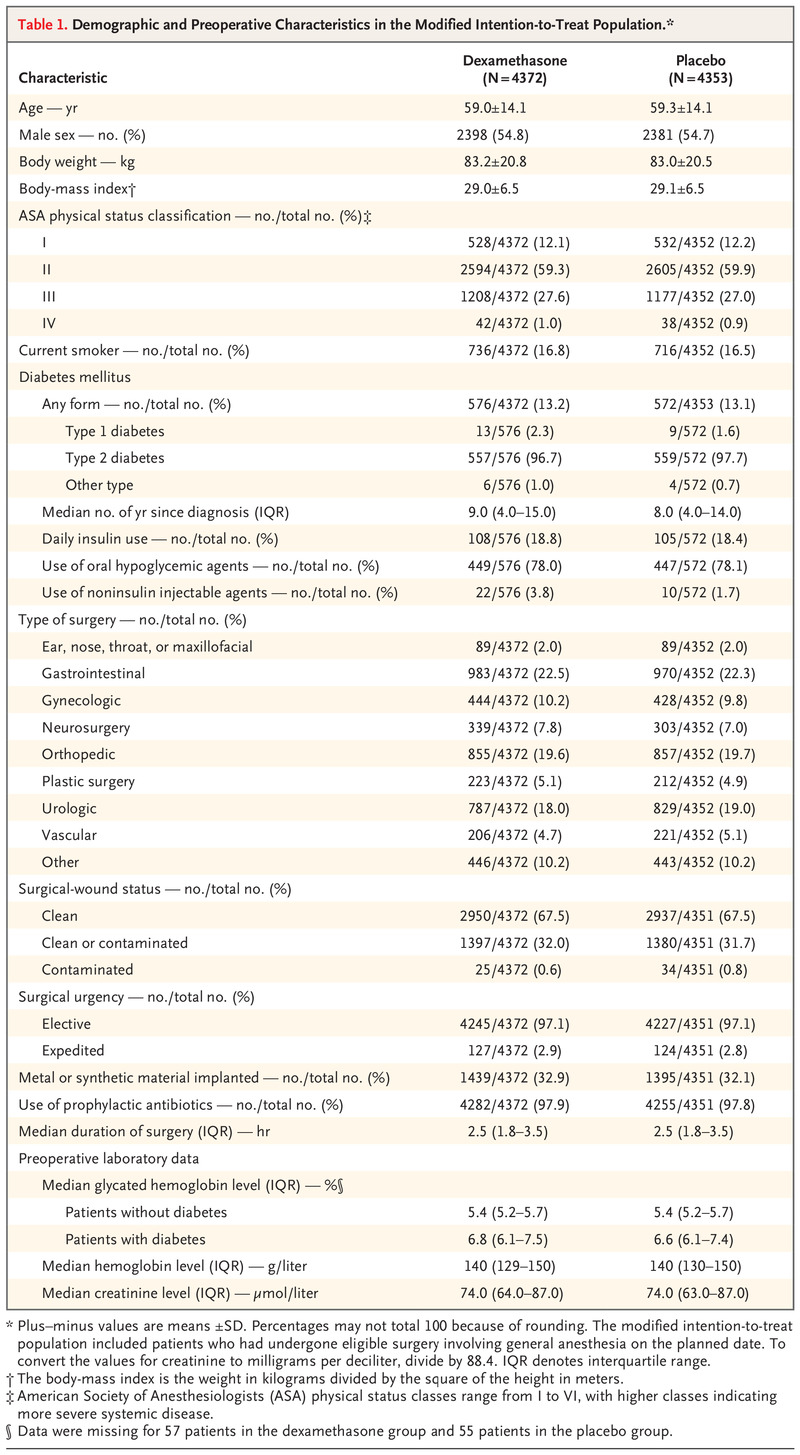
Recruitment began on March 10, 2016; the last patient underwent randomization on July 29, 2019, and the final follow-up was completed on February 26, 2020. Of the 26,909 patients who met eligibility requirements, 8880 agreed to participate and underwent randomization. A total of 4444 patients were assigned to receive dexamethasone and 4436 to receive placebo. Of these patients, 8725 (98.3%) met the criteria for inclusion in the modified intention-to-treat population (4372 in the dexamethasone group and 4353 in the placebo group) (Figure 1). Of these 8725 patients, 1148 (13.2%) had diabetes mellitus, and 1116 (97.2%) had type 2 diabetes (Table 1).
The median number of patients per site was 98 (range, 2 to 937). The list of sites and details of their recruitment are provided in Table S3. In the modified intention-to-treat population, 30-day data were available for 8678 of 8725 patients (99.5%). There were no clinically important differences in baseline or intraoperative characteristics between the two groups (Table 1 and Tables S4 through S8). Of the patients in the modified intention-to-treat population with 30-day data, 276 of 4350 patients (6.3%) in the dexamethasone group and 286 of 4328 (6.6%) in the placebo group had protocol deviations (Figure 1).
PRIMARY OUTCOME
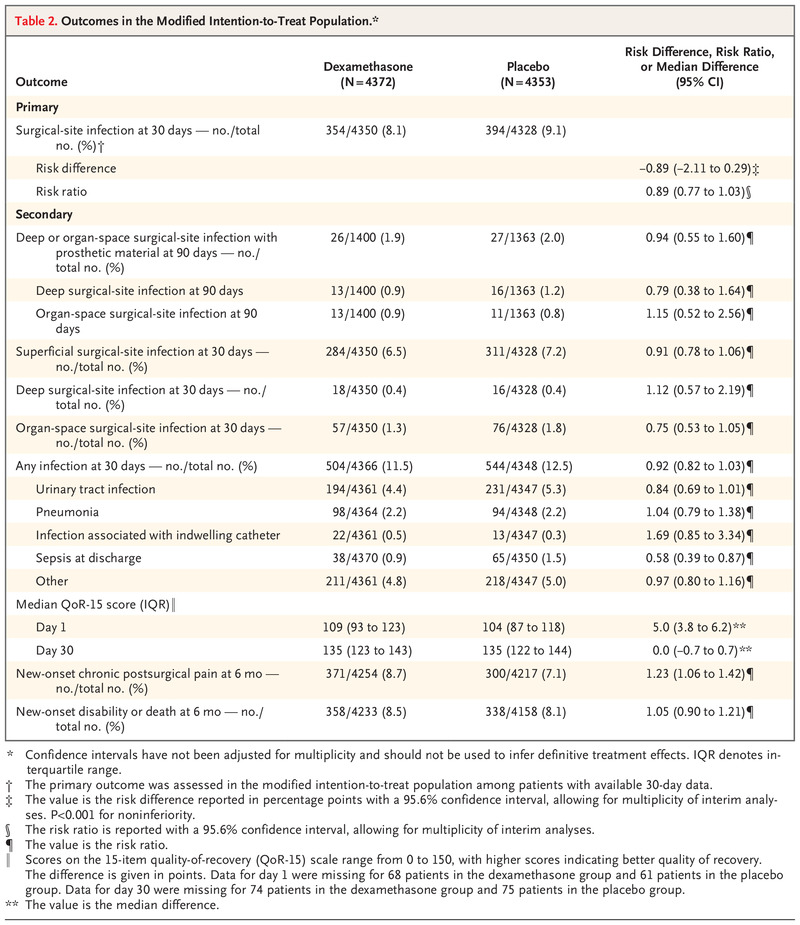
In the modified intention-to-treat population, surgical-site infection within 30 days after surgery occurred in 354 of 4350 patients (8.1%) in the dexamethasone group and in 394 of 4328 (9.1%) in the placebo group (risk difference adjusted for diabetes status, −0.9 percentage points; 95.6% confidence interval [CI], −2.1 to 0.3), a result consistent with noninferiority (risk ratio, 0.89; 95.6% CI, 0.77 to 1.03; P<0.001 for noninferiority) (Table 2). The results of the primary analysis were consistent with noninferiority in the per-protocol population (risk difference, −0.9 percentage points; 95.6% CI, −2.1 to 0.3; P<0.001 for noninferiority) and in the as-treated population (risk difference, 0.04 percentage points; 95.6% CI, −1.2 to 1.2; P=0.001 for noninferiority) (Figs. S1 and S2). These results differed minimally in sensitivity analyses with imputation for missing data and after adjustment for trial site (Tables S10 and S11).
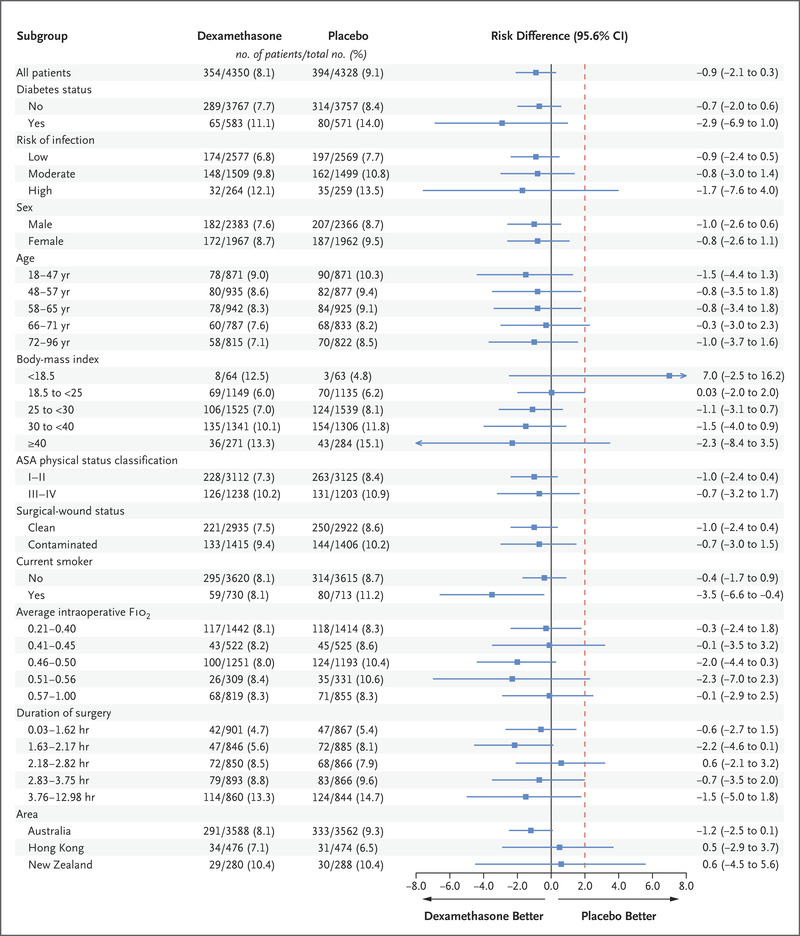
The effect of dexamethasone on the incidence of surgical-site infection was consistent across all prespecified subgroups (Figure 2). Noninferiority of dexamethasone to placebo was shown in patients with or without diabetes (risk difference in patients with diabetes, −2.9 percentage points [95.6% CI, −6.9 to 1.0]; risk difference in patients without diabetes, −0.7 percentage points [95.6% CI, −2.0 to 0.6]). There was also no compelling evidence of heterogeneity according to the presence or absence of diabetes or according to other subgroups in the per-protocol or as-treated populations (Figs. S1 and S2).
SECONDARY OUTCOMES
Secondary outcomes are shown in Table 2. The incidences of superficial, deep, or organ-space infections assessed individually were similar in the two groups. The incidence of deep or organ-space surgical-site infections within 90 days after surgery in patients with implanted prosthetic material was also similar in the two groups, when considered separately or together. Sepsis that had occurred by the day of discharge was observed in 0.9% of patients in the dexamethasone group and in 1.5% of patients in the placebo group (risk ratio, 0.58; 95% CI, 0.39 to 0.87). The median QoR-15 scores on day 1 were 109 and 104, respectively (median difference, 5.0 points; 95% CI, 3.8 to 6.2). Chronic postsurgical pain at 6 months after surgery was observed in 8.7% of patients in the dexamethasone group and in 7.1% in the placebo group (risk ratio, 1.23; 95% CI, 1.06 to 1.42).
TERTIARY AND SAFETY OUTCOMES
Discussion
In this large, pragmatic, noninferiority trial involving patients undergoing nonurgent, noncardiac surgery of 2 hours or more in duration while the patients were under general anesthesia and involving an overnight hospital stay of at least 1 day, a single 8-mg dose of dexamethasone was found to be noninferior to placebo with respect to the primary outcome of surgical-site infection within 30 days after surgery. This finding was consistent across all prespecified subgroups, including patients with or without diabetes mellitus, and held true for the individual subtypes of surgical-site infections.
Long-term glucocorticoid therapy is associated with an increased risk of surgical-site infection and wound dehiscence.18-19 There has therefore been concern that a single intraoperative dose of dexamethasone could influence the risk of surgical-site infections because it has a biologic half-life of up to 72 hours20 and leads to innate immune-cell gene expression and activation effects.5 Previous trials have not shown an increase in the risk of infection associated with the use of dexamethasone.21-23 These trials either used multiple doses of dexamethasone and examined composite end points23 or administered a single intraoperative dose that was smaller than that used in our trial.21 Some trials also excluded patients with diabetes mellitus22 or did not examine the risk of surgical-site infection as the primary outcome.21-23 We assessed a single 8-mg dose of dexamethasone because this dose is commonly used in practice and has additional analgesic benefits over the 4-mg dose when used as an antiemetic.1,3
The percentage of patients with surgical-site infection at 30 days in this trial involving patients undergoing noncardiac surgery was 8.1% in the dexamethasone group and 9.1% in the placebo group. These results are consistent with the findings in a placebo-controlled, randomized trial that assessed the effects of a larger dose of dexamethasone in patients undergoing cardiac surgery. In that trial, the incidence of wound infection appeared to be similar in the two groups; however, the overall risk of all postoperative infections was lower in the dexamethasone group than in the placebo group.24 Our observation that the results in the subgroup of patients with diabetes mellitus were similar to those of the primary analysis is reassuring, since patients with diabetes are at a higher risk for complications related to infection and for hyperglycemia, and there is therefore reluctance to use dexamethasone in patients with diabetes.3,7 The percentage of patients with diabetes in this trial (13.2%) was at the lower end of previous reports in patients who had undergone noncardiac surgery (14 to 19%).25,26 This may be because of the lower prevalence of diabetes in our population than in the predominantly North American populations in previous trials.27
Limitations of our trial warrant attention. Nonadherence to the assigned treatment may bias the analyses in the modified intention-to-treat population toward noninferiority. However, nonadherence was low and was similar in the two groups (6.3% in the dexamethasone group and 6.6% in the placebo group). Nonadherence was largely explained by the administration of supplemental nontrial glucocorticoids in the postoperative period, which underscores the common use of postoperative glucocorticoids in practice for wound-related issues such as ongoing pain or swelling. The analyses in the per-protocol and as-treated populations similarly supported the noninferiority of dexamethasone, although these results do not represent comparisons of trial groups according to randomized assignment. In addition, in this pragmatic trial, we did not collect data on perioperative factors that might influence the risk of surgical-site infection, such as subtypes of gastrointestinal surgery, the types of preoperative bowel and skin preparation, or wound treatment and details of perioperative antibiotic prophylaxis (drug class and duration of treatment). In a trial of this size, however, we would expect these factors to be evenly distributed between the groups. It is possible that the postoperative surveillance may have missed some mild infections among patients who did not return for further treatment, but we would expect such events to have been uncommon and unlikely to have altered our findings.
We found no evidence of differences between the groups in the prespecified safety outcomes. Higher differences in the blood glucose level between the preoperative blood glucose value and the maximum value in the dexamethasone group and a higher incidence of hyperglycemic events and insulin use in patients without diabetes are expected consequences of treatment with dexamethasone. The apparent increase in the incidence of new-onset chronic postsurgical pain is an unexpected finding that has not been identified in previous studies.28 We did not adjust for multiplicity, and this finding may be explained by chance. Future analyses will further assess pain and disability outcomes. Tertiary analyses supported reductions in the risk of nausea and the use of antiemetics in the first 24 hours after surgery associated with dexamethasone, a finding consistent with previous reports.1-3,22
Supported by a grant (APP1079501) from the Australian National Health and Medical Research Council (NHMRC), a grant (GRF 14101816) from the Research Grants Council of Hong Kong, and Monash University. Dr. Corcoran was supported by a Health Department of Western Australia Raine Foundation Clinical Practitioner Fellowship, Dr. Myles by an NHMRC Practitioner Fellowship (GNT1135937), and Dr. Cheng by an NHMRC Career Development Fellowship.
Author Affiliations
From Royal Perth Hospital (T.B.C., P.C., K.M.H.), the University of Western Australia (T.B.C., E.O., K.M.H.), Murdoch University (K.M.H.), and Fiona Stanley Hospital (E.O.), Perth, and the Alfred Hospital (P.S.M., A.C.C., L.A.B.), Monash University (T.B.C., P.S.M., A.B.F., A.C.C., L.A.B., K.L., C.M.), the University of Melbourne (K.L., D.S.), and Royal Melbourne Hospital (K.L.), Melbourne, VIC — all in Australia; the Chinese University of Hong Kong, Hong Kong (M.T.V.C.); and Auckland City Hospital and the University of Auckland — both in Auckland, New Zealand (T.G.S.).
Address reprint requests to Dr. Corcoran at the Department of Anaesthesia and Pain Medicine, Royal Perth Hospital, Wellington St., Perth, WA 6000, Australia, or at tomas.corcoran@health.wa.gov.au.
Participating centers and investigators in the PADDI trial are listed in the Supplementary Appendix, available at NEJM.org.